


Why do Wu et al. 2020 refuse to answer scientific questions regarding the claimed SARS-CoV-2 sequence?
Several attempts to contact the original authors remain unanswered, even via Prof. Holmes.

Background
In my genomics series, I have tried to unravel, what happened in late 2019 and early 2020, as the team around Prof. Zhang and the Chinese CDC identified the first sequence claimed to be the genome of SARS-CoV-2.
To this date, several of us have not been able to reproduce the exact contigs used to produce the current reference genome of the first SARS-CoV-2 genome variant.
Also, nobody has to date shown that the ends of the genome can be aligned perfectly with multiple reads, thus showing that the genome itself exists as a dedicated entity.
Overview of my Seven Part Genomics Series:







Twitter Debate with McKernan
In a recent Twitter debate, Kevin McKernan said, that the reads would have to be trimmed by the four (or even five as shown in the red box) random bases, that were added by the Takara protocol, in order to properly align them to the head of the genome.
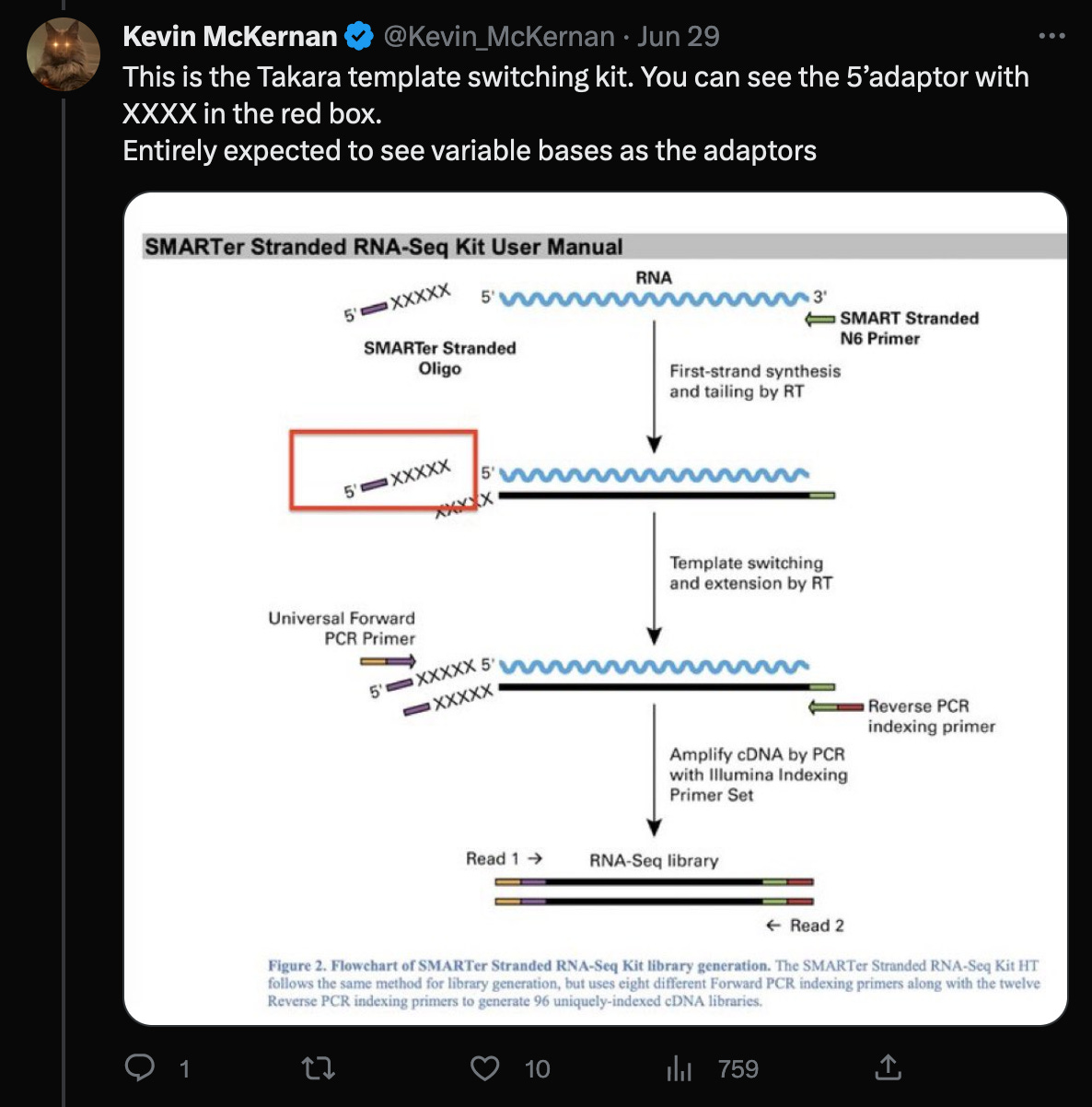

However, further investigation of these claims reveal:
The statement of trimming of four nucleotides, invalidates McKernan’s earlier statement, that adaptors would not ligate (bind) to the ends of the genome in the first place. (Of course, that’s why the random adaptors are used, but it would still make the statement irrelevant, as this would not be a problem in this case.)
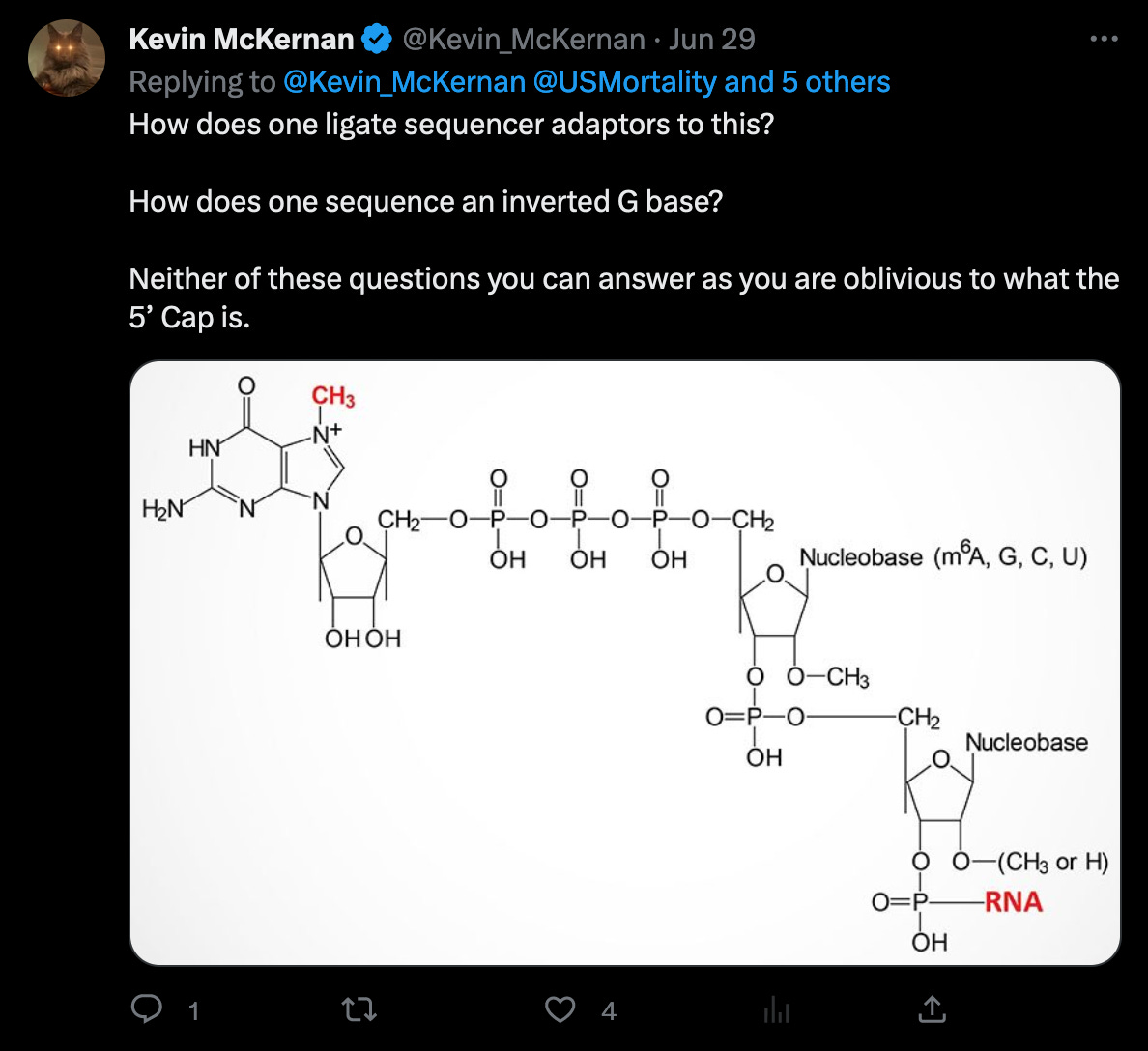
According to the Takara manual, there are three nt’s not four (!!) after the adaptor sequences, that must be trimmed.

McKernan’s screenshot omits the full length of the reads, which shows, that there are only eleven reads (of the 124 alleged coverage), that would perfectly align, when clipping the first four bases - but of course, according to the Takara manual, it should be three not four bases that are being clipped after all!
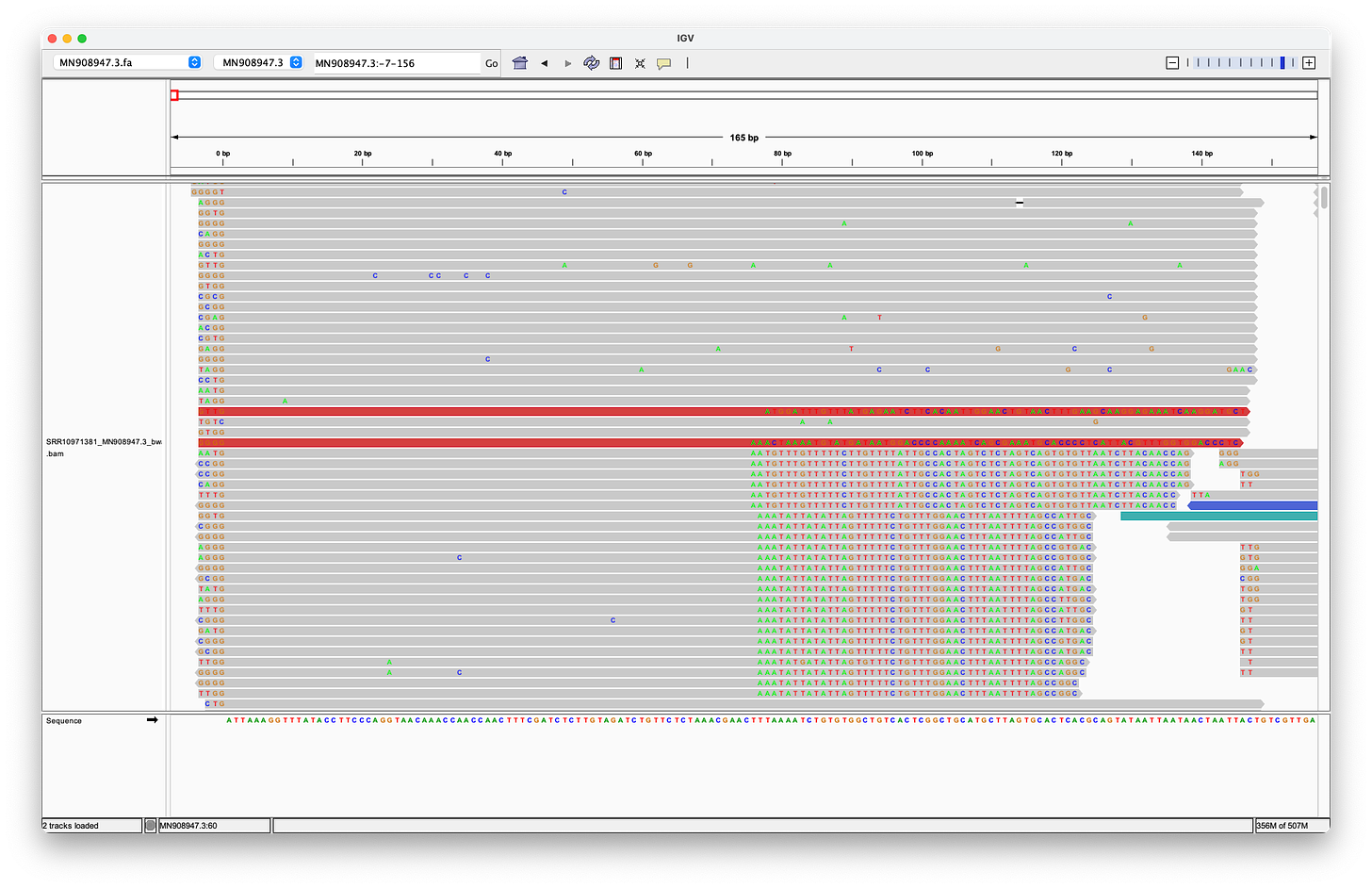
./src/align.sh --sra SRR10971381 --genome MN908947.3 --aligner bwa Even when clipping off three bases, we get exactly two reads, that perfectly align to the head. But we still don’t get any matches at the end! (End not shown)


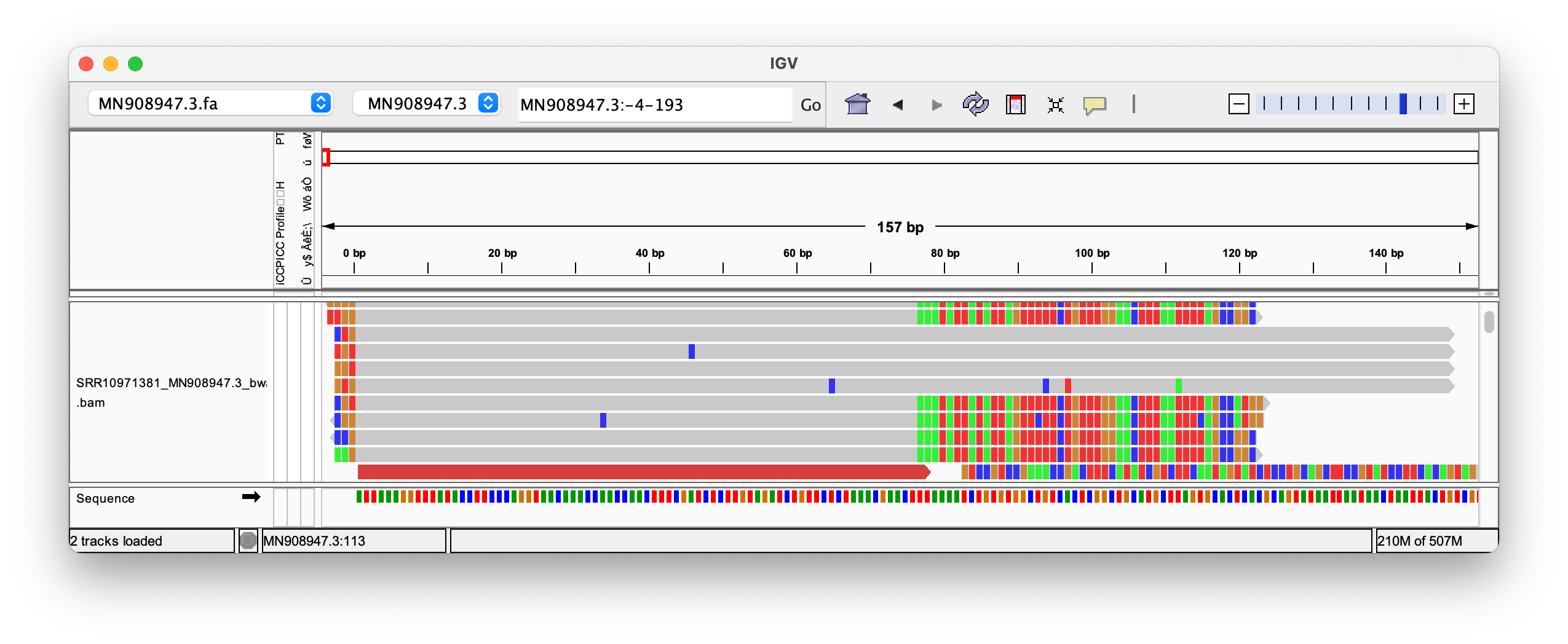
Even when trimming off the leading three or four nucleotides from the reads, Megahit still assembles de-novo the head with two extra G bases at the head!
Wu et al. 2020, wrote:

They say "56.5M reads" were remaining, after trimming. This matches exactly the count of the uploaded SRA. When quality trimming with trimmomatic, this typically leads to some reads being dropped. Thus, it can be inferred, that the uploaded SRA has already been trimmed.
Also, one can tell that the adaptors were probably trimmed, from the way that there are several reads which match the Illumina TruSeq single index adaptors apart from one or two errors. So the errors probably prevented the adaptors from getting trimmed.This is strong evidence, that the uploaded SRA might be already adaptor trimmed.
There are no regular adaptor sequences to be found in the SRA. So why would Wu et al. trim the adaptor sequences, but leave the three random bases? The Miniseq manual states: “Local Run Manager for MiniSeq will automatically perform adaptor trimming for built-in Illumina kits only.” It remains unclear if the operators had turned off the automatic adaptor trimming or not.

Are the adaptors trimmed or not?
As you can from the above six bullet points, it is still not possible to align the head and the tail perfectly with the provided reads, even when assuming trimming of 3 or 4 bases. It is also unclear if the uploaded dataset was already adaptor trimmed or not. Evidence shows, that the uploaded SRA might be already trimmed. In order to clarify, I’ve tried to reach out to the authors of Wu et al!

I’ve received no reply from any of the authors, except the only non-Chinese, Prof. Holmes from Australia. Here’s my e-mail correspondence (personal info redacted), where I ask three technical questions:
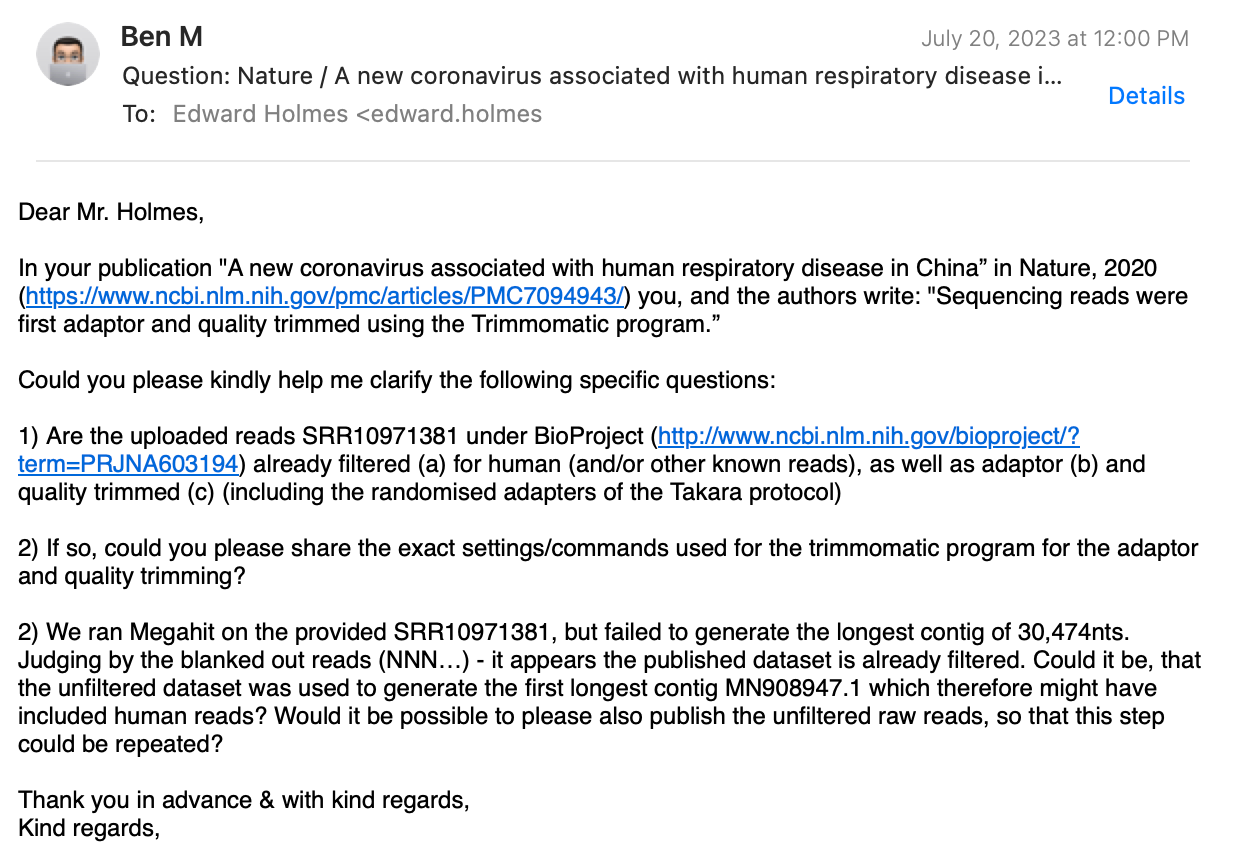
Prof. Holmes replied, stating that only Prof. Zhang and his team did the bioinformatics work, which implies that he is not aware of the answers to my questions!
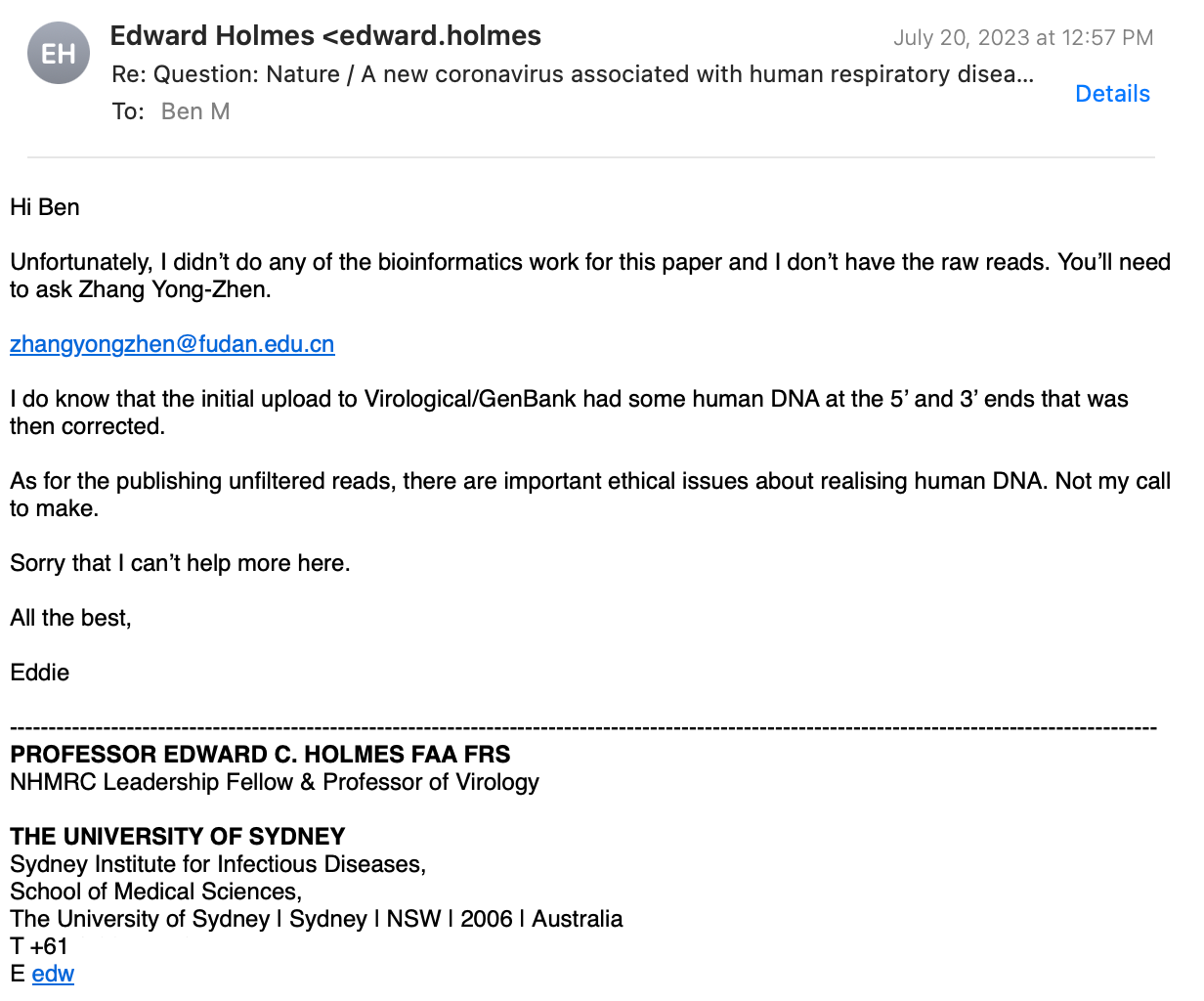
I followed up, explaining that none of the Chinese authors, including Prof. Zhang replied. I did not receive any message.
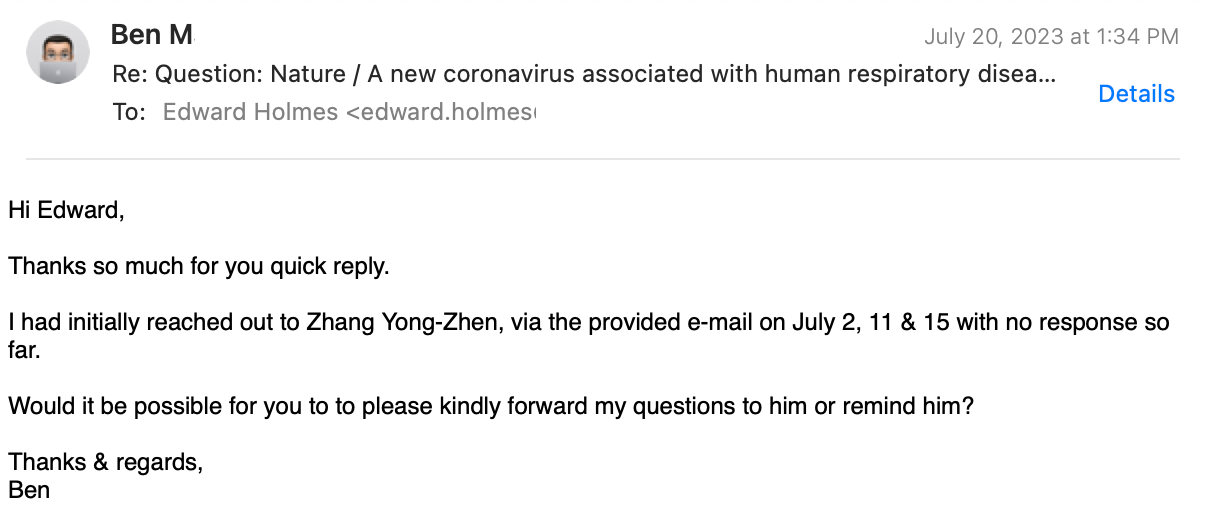
Prof. Holmes replied again:

I had to ask again:
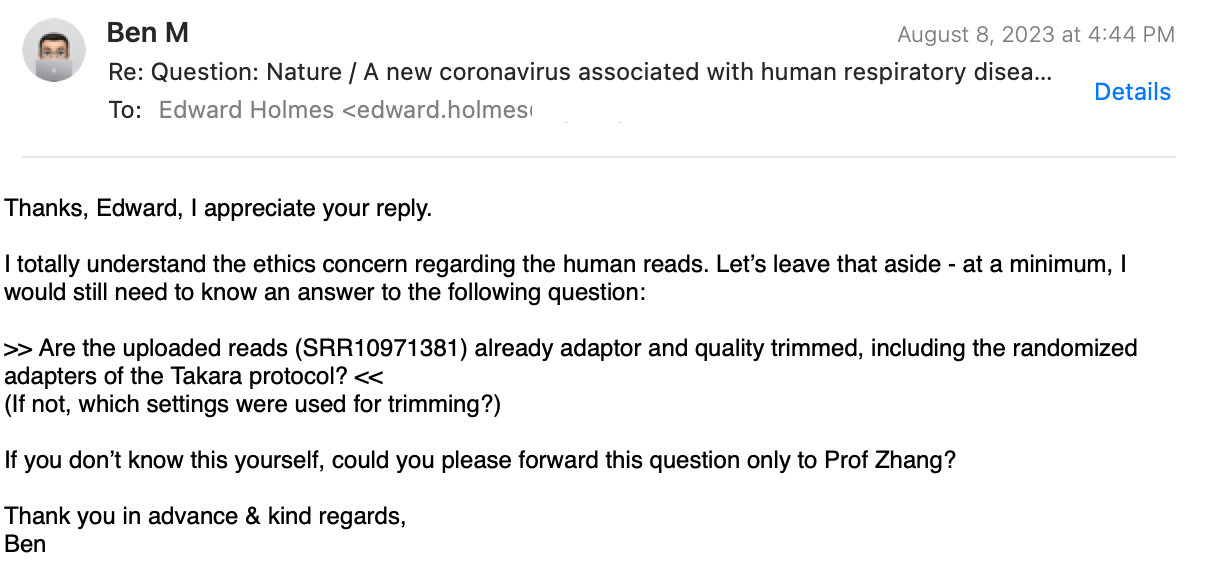
Finally, he agrees, to ask the Chinese for me!

After one week had passed again, I followed up:
And his final answer:

Hence, unfortunately my correspondence with the original authors provided no insights into the matter, due to lack of willingness or knowledge.
So why does the co-author of the paper, Prof. Holmes, Professor of Virology, not know the answers to my questions
- and -
Why do the Chinese remain silent?
Nanopore Reads
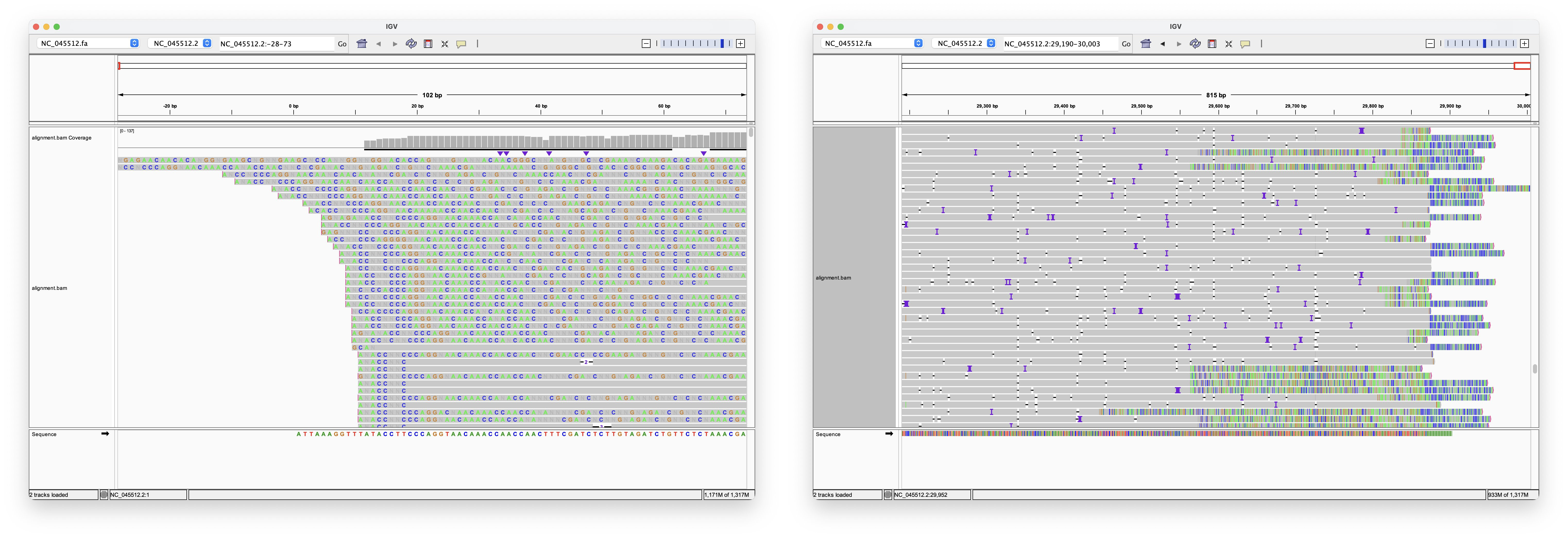
Nanopore can produce much longer reads than the ~150bp used in the Wu et al. paper, and is often presented as the ultimate proof for the validity of the sequence.
Hence, I have used the reads provided in this paper, to align them. As you can see, the quality of these long Nanopore reads is really terrible - so many errors and inserts! I could not find a single read that perfectly aligns to either the head or the tail! Note, that the highlighted letters are errors, and the purple/black areas are inserts/deletions. Judge for yourself, but in my opinion, the amount of errors, does not increase confidence in the genome.
Update (12/11/2024):
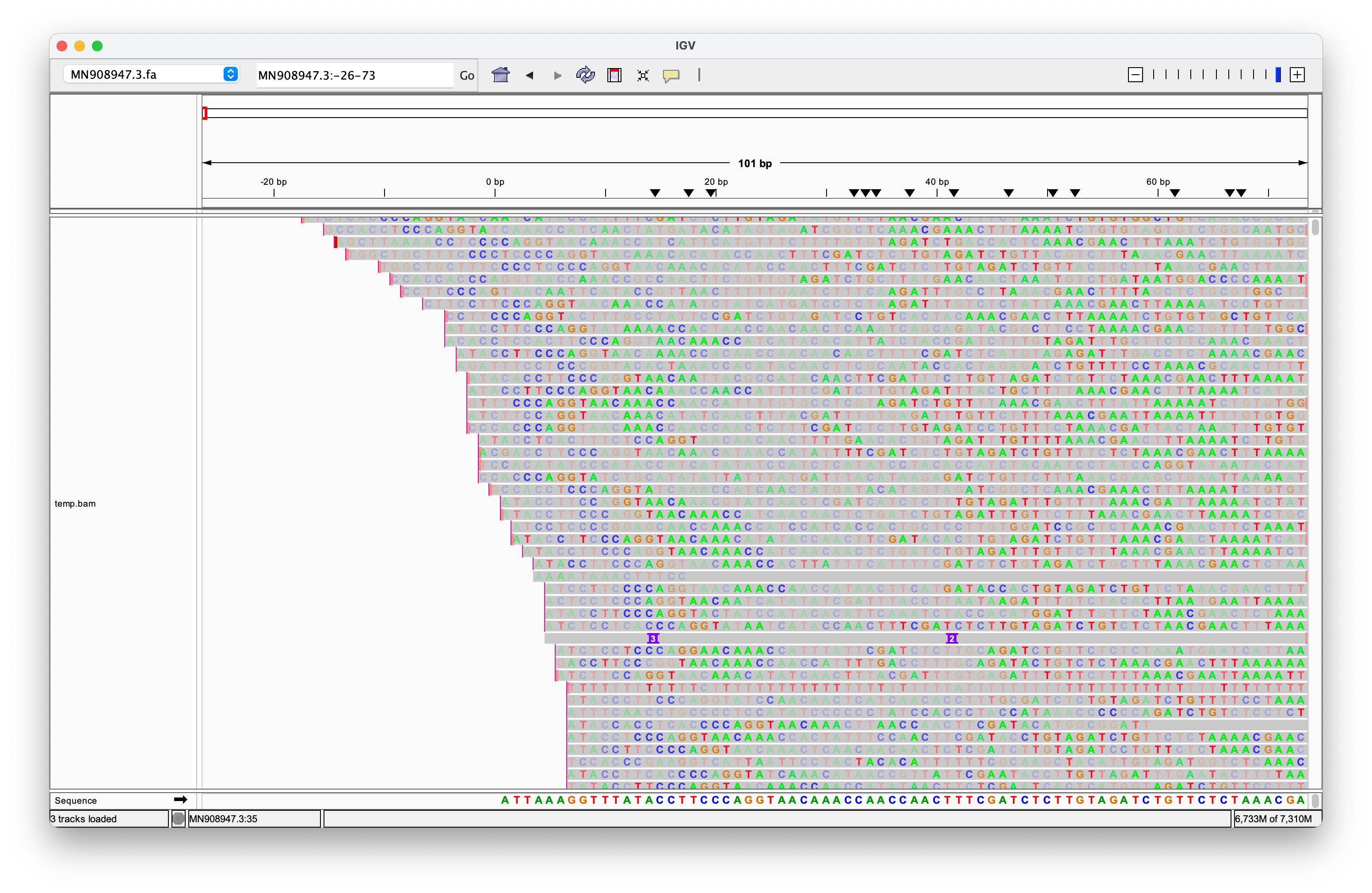
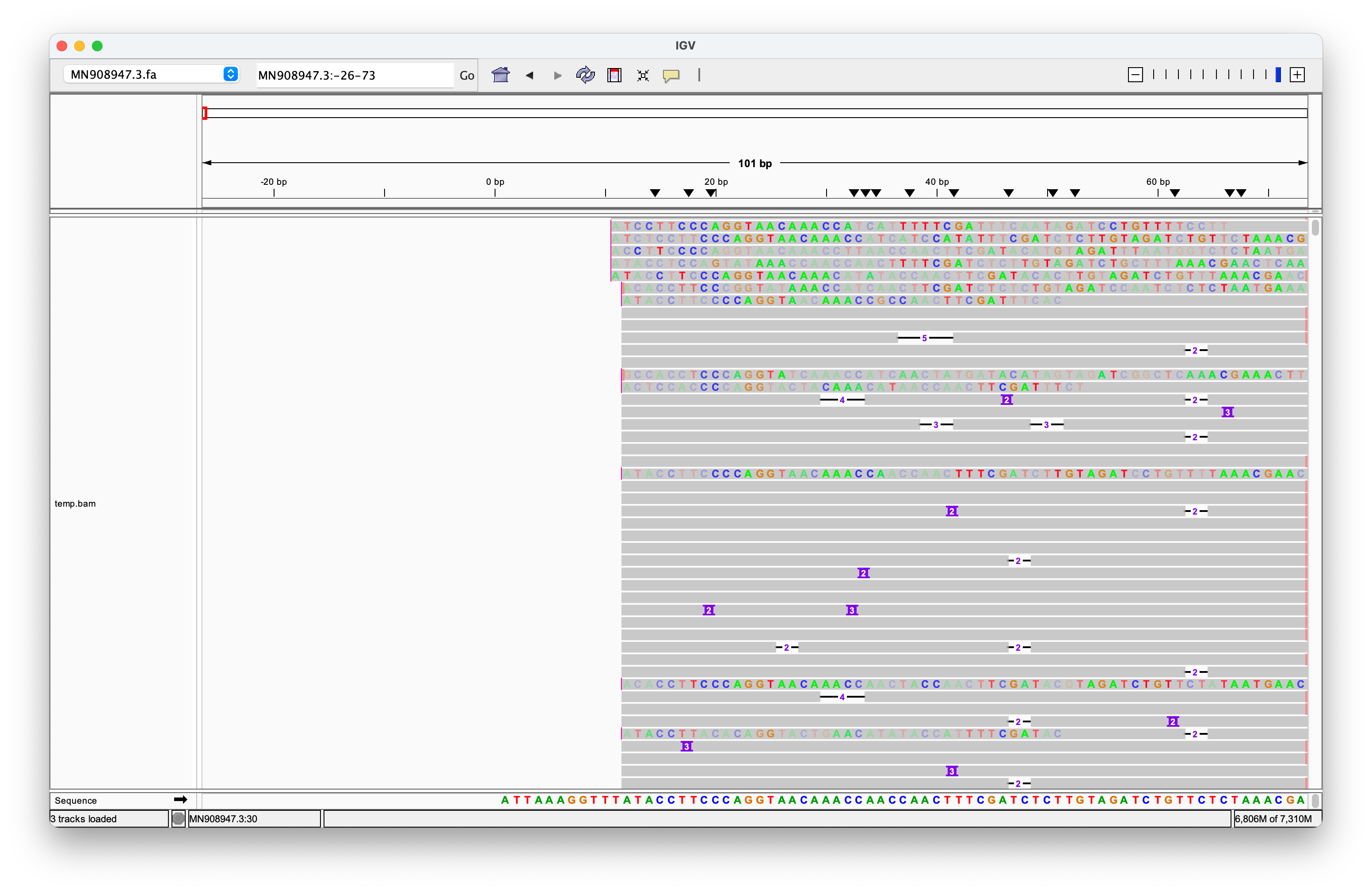
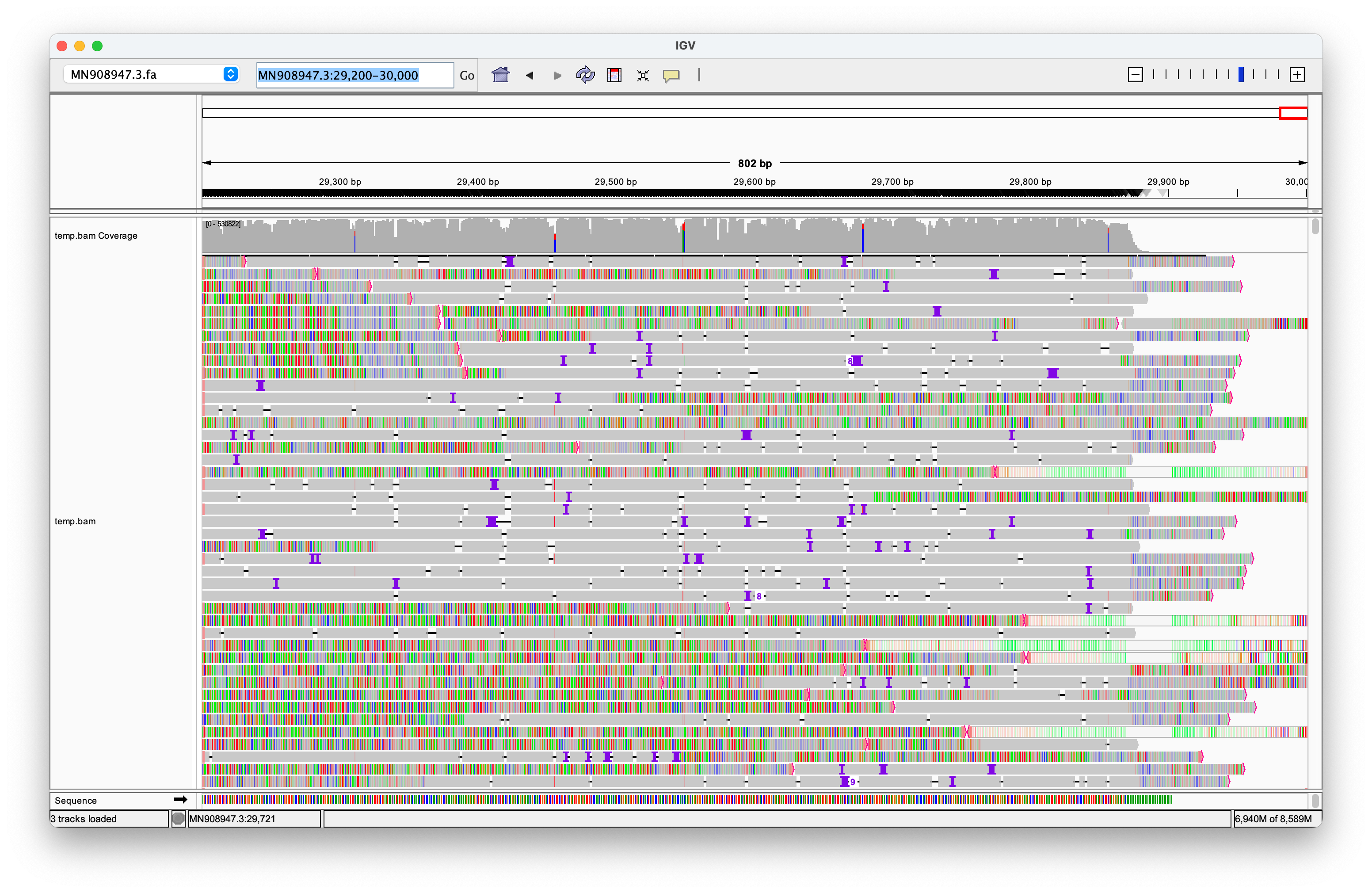
McKernan and Henjin have repeatedly used the screenshot above to launch further attacks. Their critique centers on my use of the DNA SRA version instead of the directly sequenced RNA version, and my subsequent oversight in not converting it prior to alignment. However, this argument is merely a distraction from the core issue: The Nanopore reads do not exhibit robust alignments to the ends of the genome. Moreover, many reads that do align near the ends often include longer sequences of unrelated genetic material before or after, strongly suggesting that they do not originate from a distinct single-stranded RNA genome.
Additionally, I included the clipped portions of the reads, which only reinforces my argument. It is illogical for reads to require clipping to align with a genome strand if they truly originate from that strand.
I have left the original screenshot intact as evidence of my commitment to pursuing the truth. I am always open to improving my work and I am willing to acknowledge and correct errors. However, the language and personal attacks from individuals like McKernan and others raise significant concerns about their intentions.
Conclusion
1) No one, to this date, was able to perfectly reproduce MN908947.3 with Megahit or any other assembler, even when trimming the alleged adaptors, we still end up with two (or three) leading G's and a missing tail.
2) There are no reads in the sample which match perfectly. At best, there are four reads that map well to the head - when you assume trimming three bases is accurate, which you don't know for a fact. There's none for the tail.
My initial requirements, lined out in https://usmortality.substack.com/p/is-the-sars-cov-2-genome-valid have NOT been fulfilled, which were:
1) At least 10–20 reads that show a perfect alignment to the head & the tail.
2) A complete genetic sequence (RNA) of ~30kb length exists in the sample.
Obviously, a valid sequence assembly would still need to be shown to originate from the virus particle (thus be a genome), and then demonstrate causation of illness. The latter, should be fairly easy, if you can present a study, i.e. a double blinded RCT e.g. in an animal model where a synthesized sequence leads to production of the same particle and to uncontrolled spread and disease via subsequent natural exposure.
How many virions were in the original sample? Surely, if the patient had pneumonia caused by the virus, it must've been thousands or even more?
I have already shown that 100 virions, randomly fragmented, lead to about 26 matches of which maybe half are perfect, in a simulated environment. Hence, thus far, there's no conclusive evidence, that there were such virions in the sample.
Summary
I was unable to receive a reply from the team around Wu & Zhang.
Prof. Holmes has no knowledge of the requested information, and was not able to retrieve it from the Chinese either.
McKernan’s statements of requiring four bases to be trimmed off, is likely wrong, as the Takara manual requires three bases, remains speculative in nature, and additional evidence shows, that the uploaded SRA was likely adaptor trimmed already.
Even with the speculative trimming of three or four bases, only four to eleven reads align perfectly to the head and none to the tail, while Megahit still fails to produce a head with two extra G bases.
Nanopore reads show no alignments of head/tail & low quality alignments along the middle of the genome.
This means, that in my opinion, the validity of the original SARS-CoV-2 genome is still not scientifically and accurately established, as it should be possible to multiple reads that align perfectly against both ends, as shown in my simulated read experiment!
Please let me know your thoughts!
Thank you for this very important work! Can you lay out, perhaps here or perhaps in an article of its own, a walk through for lay people on this issue? Have you done that in a past piece? Similarly, can you lay out the scenario of what the assembly assembled, if it was NOT a new virus? Another questions, the CEO of Illumina is on record saying "his team" was flown into China to do the first sequence. Thoughts on that?
To Australians in the know, Prof. Ed Holmes is known as 'sweaty Eddie.'
Sir was on TV multiple times in 2020 discussing the origins of covid, looking like someone was holding a gun to his head off screen sweating profusely. It's not about your emails, and it is definitely not about science. It's about the US DOD, the ADF, WHO and the CCP. He has been given his instructions. Tread lightly.
Edit: Hope that answers your question about why he told you to FO and why no one in China will contact you.