


SARS-CoV-2 Genome Assembly
An attempt to regenerate the claimed SARS-CoV-2 genetic sequence.

Here I’m trying to reproduce how the first scientists have identified and sequenced the SARS-CoV-2 Coronavirus. This is the first article of a series of three articles, so please stay tuned, share and subscribe!
Overview of my Seven Part Genomics Series:







Background
The first note about the new virus was published by Prof. Zhang (China) via the forum account of Prof. Holmes (Australia) on January 20, 2020.
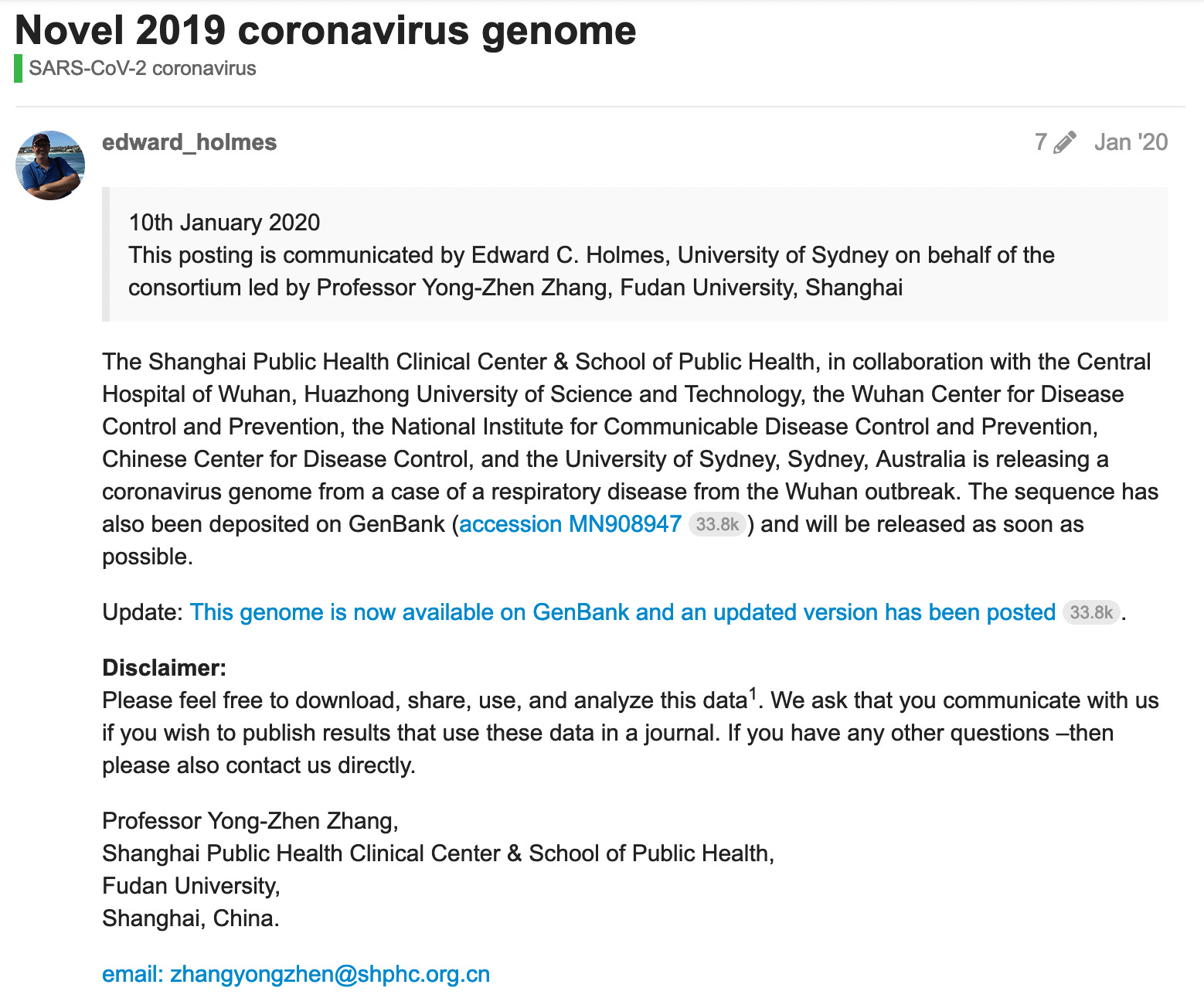
https://virological.org/t/novel-2019-coronavirus-genome/319
Q: Why did Prof. Zhang use Prof. Holmes’ account - or why did latter publish formers statement?
Noteworthy, that they have uploaded three versions of the claimed sequence by now. The first version, that was uploaded on January 14, 2020 contained 30,473 basis pairs (bp). Three days later they updated it with a shorter version with 29,875bp and on 3/18/2020 it was extended again to 29,903 bp.

https://www.ncbi.nlm.nih.gov/nuccore/MN908947
The forum entry that was posted on 1/17 points to the second version:
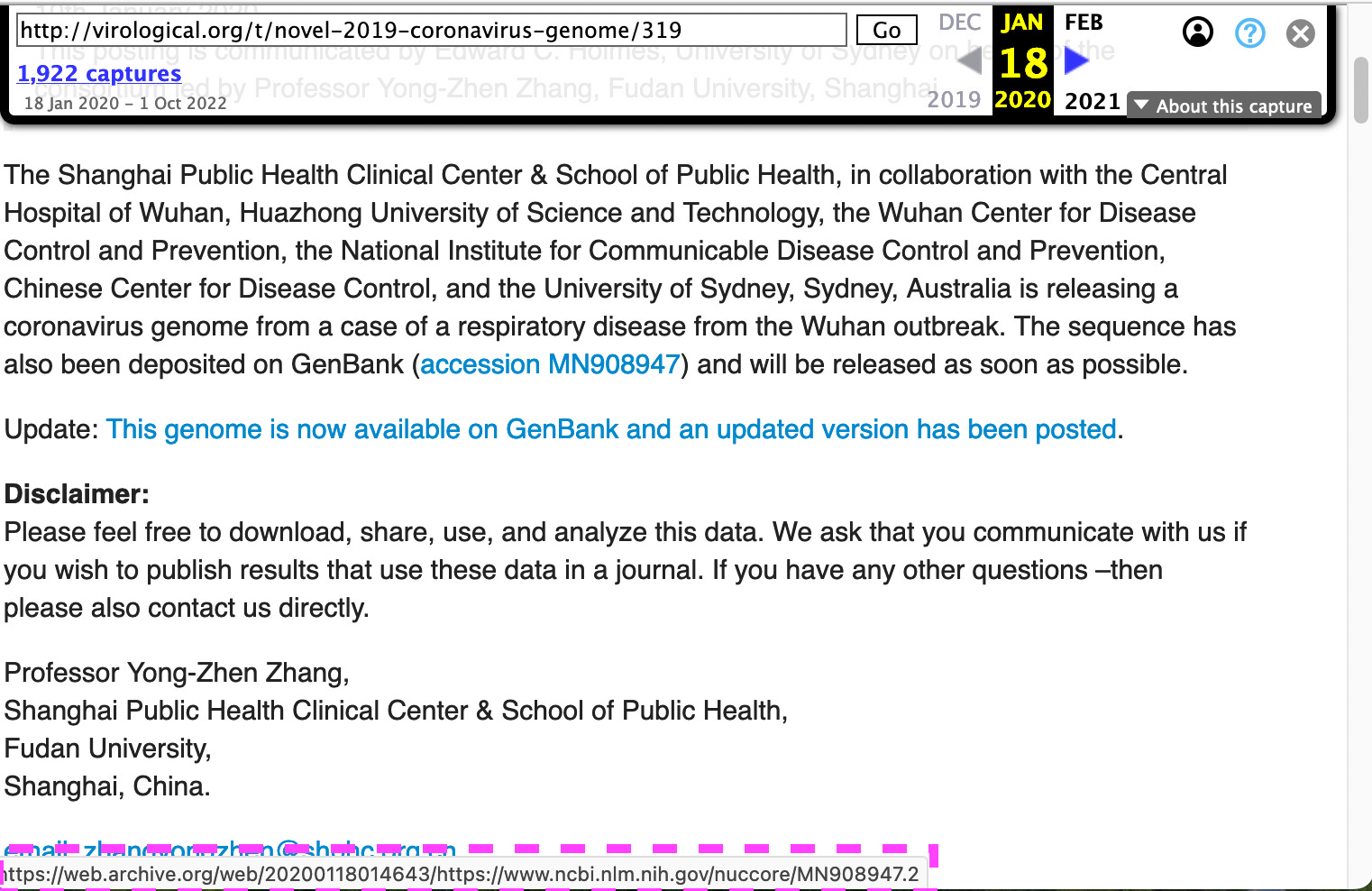
https://web.archive.org/web/20200118014643/http://virological.org/t/novel-2019-coronavirus-genome/319
So far, I have not found any information why the genome has been updated at least three times.
Q: Why was the claimed genome updated so many times?
Zhang & Wu have published their accompanying paper for the sequence here:

https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7094943/
Here they describe how they have taken a sample of a “41-year-old man”, with the following symptoms: “fever, chest tightness, unproductive cough, pain and weakness for 1 week”. He was administered several medications, but still had to be put on a ventilator in the ICU. They mention that later he was moved to another hospital - which is wild, I’m not sure why people that are on a ventilator and in an ICU are moved between hospitals. The outcome of the patient remains unknown from the paper.
Q: Why was the patient moved between hospitals while being in ICU on a ventilator, and what was his final outcome?
Sequencing
In the paper, the authors describe how they generated the sequence.

So they took bronchial fluid from this patient, converted it to DNA and processed it with a Sequencer, which reads the genetic code of the mixture.
It is noteworthy, that only DNA can be sequenced, hence they had to convert all the RNA in the sample to DNA first. Noteworthy, that our own cells, internally, use RNA and of course consist of DNA. So after the conversion they have only DNA which originates of Human DNA, Human RNA and possibly RNA of many microbes (incl. viruses). Now in a latter step they write, that they excluded human DNA by matching it against the Human Genome - but:
Is it certain, that all that is left in the dataset is of non-human origin?

Now, the machine only reads sequences up to a length of 150bp, which means that it would never be able to read the full sequence of any virus. Instead, we get a large set of so-called reads with a length of up to 150bps.
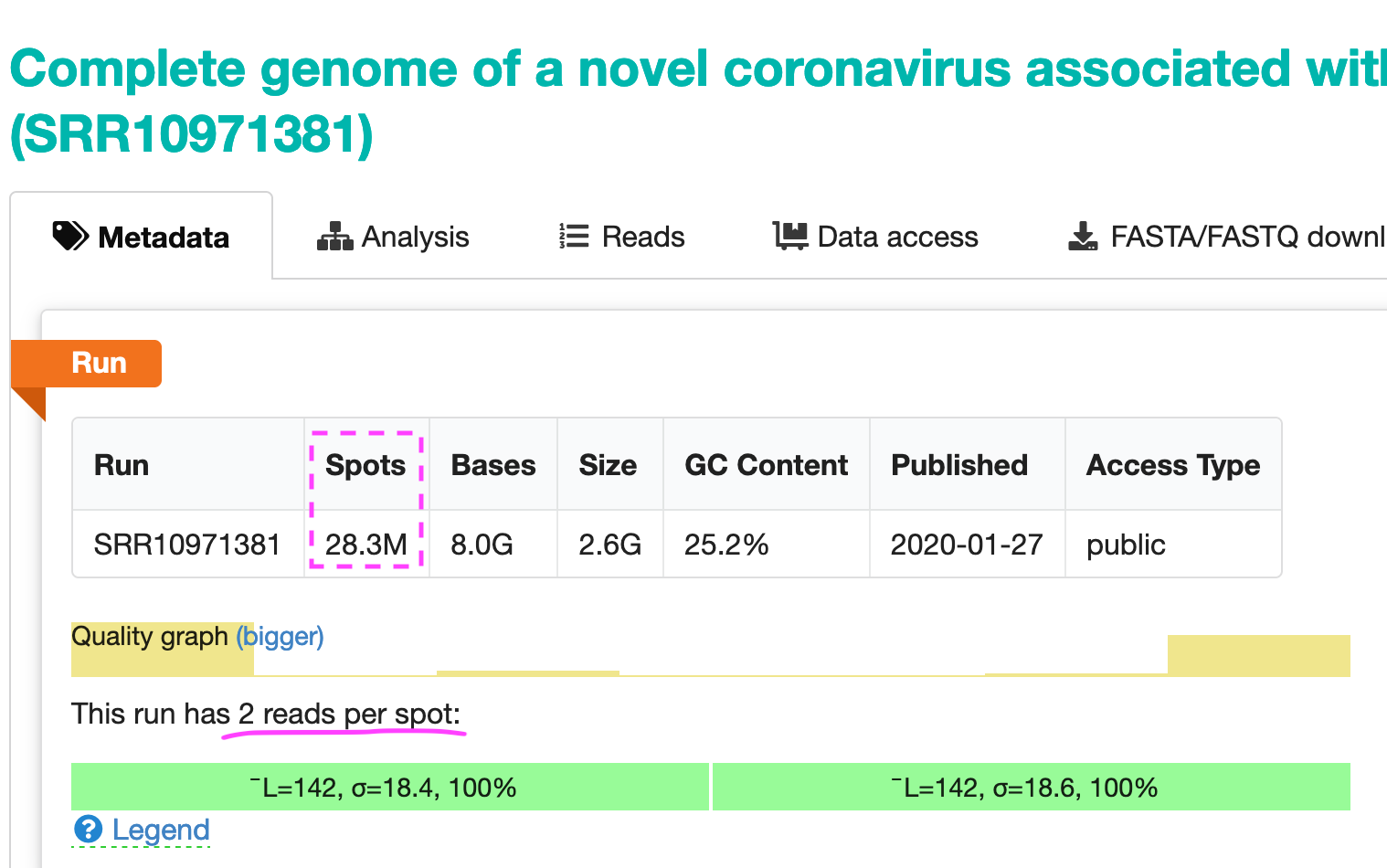
So the sequencer generated 28.3 million spots (at 2 reads per spot). They look like shown in the image below.
You can take a look at all reads here: https://trace.ncbi.nlm.nih.gov/Traces/?view=run_browser&page_size=10&acc=SRR10971381&display=reads
Assembly
Now that we know where these reads came from, let’s try to assemble them with the bioinformatic tools. I’ve previously searched on GitHub for a script that would simply run the entire assembly pipeline, but failed to find one.
So I build my own, and this is what I ended up with:
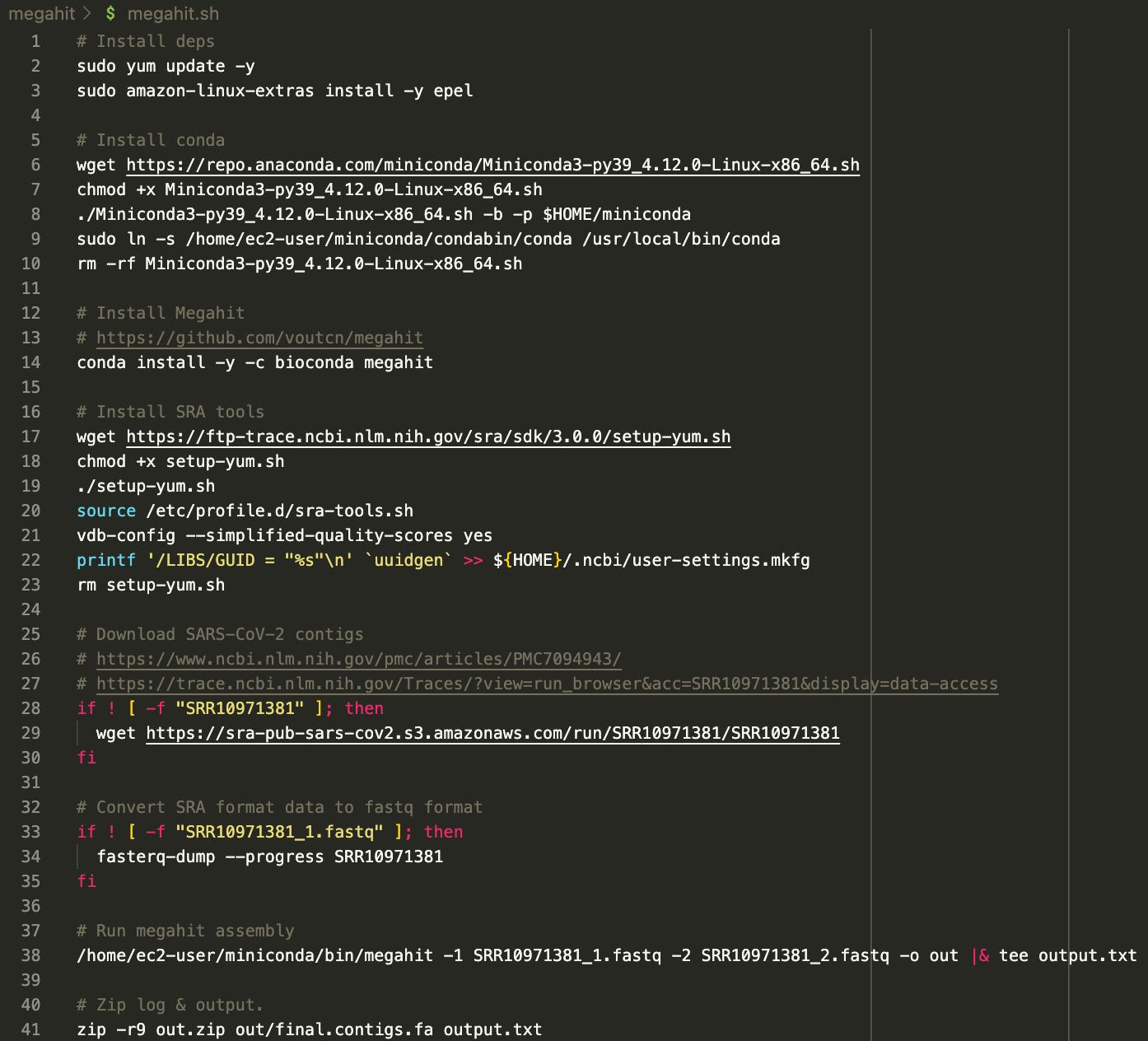
The script is available here: https://github.com/usmortality/Megahit-SARS-CoV-2
The assembly is done with a tool called Megahit. All necessary dependencies are installed, and the “reads” are downloaded, converted to fastq format and then fed into Megahit. Finally, the result is zipped.
I have ran this script on a 32-core AWS compute optimized instance as follows:
aws ec2 run-instances --image-id ami-026b57f3c383c2eec --count 1 --instance-type c5a.8xlarge --key-name ben --ebs-optimized --block-device-mapping "[ { \"DeviceName\": \"/dev/xvda\", \"Ebs\": { \"VolumeSize\": 100 } } ]"
scp megahit.sh megahit:/home/ec2-user/.
ssh megahit
chmod +x megahit.sh
screen
./megahit.shResult
Here’s the result of my run with Megahit 1.1.3
62.126Gb memory in total.
Using: 55.913Gb.
MEGAHIT v1.1.3
--- [Sat Oct 1 16:42:12 2022] Start assembly. Number of CPU threads 32 ---
--- [Sat Oct 1 16:42:12 2022] Available memory: 66706915328, used: 60036223795
--- [Sat Oct 1 16:42:12 2022] Converting reads to binaries ---
b' [read_lib_functions-inl.h : 209] Lib 0 (SRR10971381_1.fastq,SRR10971381_2.fastq): pe, 56565928 reads, 151 max length'
b' [utils.h : 126] Real: 46.1887\tuser: 35.0145\tsys: 10.3506\tmaxrss: 164224'
--- [Sat Oct 1 16:42:58 2022] k list: 21,29,39,59,79,99,119,141 ---
--- [Sat Oct 1 16:42:58 2022] Extracting solid (k+1)-mers for k = 21 ---
--- [Sat Oct 1 16:44:04 2022] Building graph for k = 21 ---
--- [Sat Oct 1 16:44:55 2022] Assembling contigs from SdBG for k = 21 ---
--- [Sat Oct 1 16:47:27 2022] Local assembling k = 21 ---
--- [Sat Oct 1 16:47:57 2022] Extracting iterative edges from k = 21 to 29 ---
--- [Sat Oct 1 16:48:52 2022] Building graph for k = 29 ---
--- [Sat Oct 1 16:49:08 2022] Assembling contigs from SdBG for k = 29 ---
--- [Sat Oct 1 16:50:17 2022] Local assembling k = 29 ---
--- [Sat Oct 1 16:50:50 2022] Extracting iterative edges from k = 29 to 39 ---
--- [Sat Oct 1 16:51:44 2022] Building graph for k = 39 ---
--- [Sat Oct 1 16:51:54 2022] Assembling contigs from SdBG for k = 39 ---
--- [Sat Oct 1 16:52:54 2022] Local assembling k = 39 ---
--- [Sat Oct 1 16:53:26 2022] Extracting iterative edges from k = 39 to 59 ---
--- [Sat Oct 1 16:54:04 2022] Building graph for k = 59 ---
--- [Sat Oct 1 16:54:12 2022] Assembling contigs from SdBG for k = 59 ---
--- [Sat Oct 1 16:54:47 2022] Local assembling k = 59 ---
--- [Sat Oct 1 16:55:22 2022] Extracting iterative edges from k = 59 to 79 ---
--- [Sat Oct 1 16:55:45 2022] Building graph for k = 79 ---
--- [Sat Oct 1 16:55:48 2022] Assembling contigs from SdBG for k = 79 ---
--- [Sat Oct 1 16:56:04 2022] Local assembling k = 79 ---
--- [Sat Oct 1 16:56:37 2022] Extracting iterative edges from k = 79 to 99 ---
--- [Sat Oct 1 16:56:54 2022] Building graph for k = 99 ---
--- [Sat Oct 1 16:56:56 2022] Assembling contigs from SdBG for k = 99 ---
--- [Sat Oct 1 16:57:03 2022] Local assembling k = 99 ---
--- [Sat Oct 1 16:57:29 2022] Extracting iterative edges from k = 99 to 119 ---
--- [Sat Oct 1 16:57:41 2022] Building graph for k = 119 ---
--- [Sat Oct 1 16:57:42 2022] Assembling contigs from SdBG for k = 119 ---
--- [Sat Oct 1 16:57:46 2022] Local assembling k = 119 ---
--- [Sat Oct 1 16:58:09 2022] Extracting iterative edges from k = 119 to 141 ---
--- [Sat Oct 1 16:58:16 2022] Building graph for k = 141 ---
--- [Sat Oct 1 16:58:17 2022] Assembling contigs from SdBG for k = 141 ---
--- [Sat Oct 1 16:58:20 2022] Merging to output final contigs ---
--- [STAT] 30425 contigs, total 14483601 bp, min 200 bp, max 29870 bp, avg 476 bp, N50 448 bp
--- [Sat Oct 1 16:58:20 2022] ALL DONE. Time elapsed: 968.493221 seconds ---https://github.com/USMortality/Megahit-SARS-CoV-2/blob/megahit-1.1.3/out/output.txt
Noteworthy, that the Wu et al. used an older version, namely 1.1.3 of Megahit from March 2018, while the latest version 1.2.9 was already available since October 2019.
Why did Wu et al. use the much older version 1.1.3 of Megahit, instead of the latest 1.2.9 version?
As to why that specific version was used remains unclear.
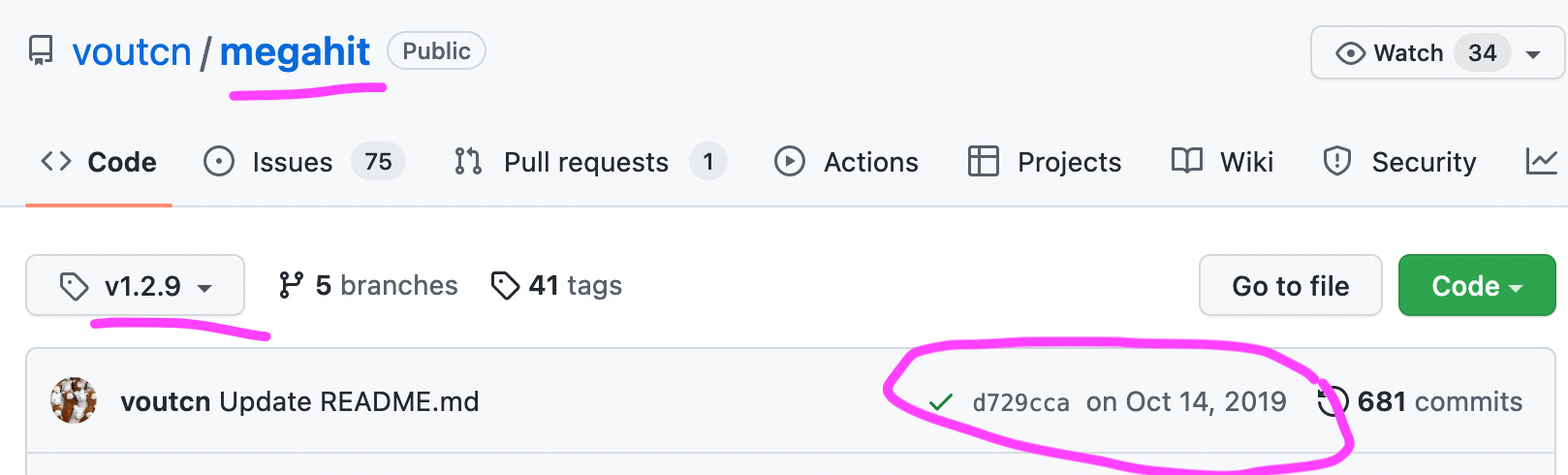
https://github.com/voutcn/megahit/tree/v1.2.9
Back to the results - we can see that assembly took about 16 minutes on a 32-core compute optimized Amazon Linux instance.
Here are the final stats:
[STAT] 30425 contigs, total 14483601 bp, min 200 bp, max 29870 bp, avg 476 bp, N50 448 bpThe authors wrote in their paper, that their assembly yielded:
Megahit generated a total of 384,096 assembled contigs (size range of 200–30,474 nt)
So while the authors assembled 384k contigs, I was only able to assemble 30k contigs (This might have to do with the additional filtering they did).
As the longest contigs generated by Megahit (30,474 nt)
Also, I am not able to regenerate the longest contig, which had over 30k bp, while my run only assembled 29,870bp.
Here are the results summarized, my runs are the two bars on the right:
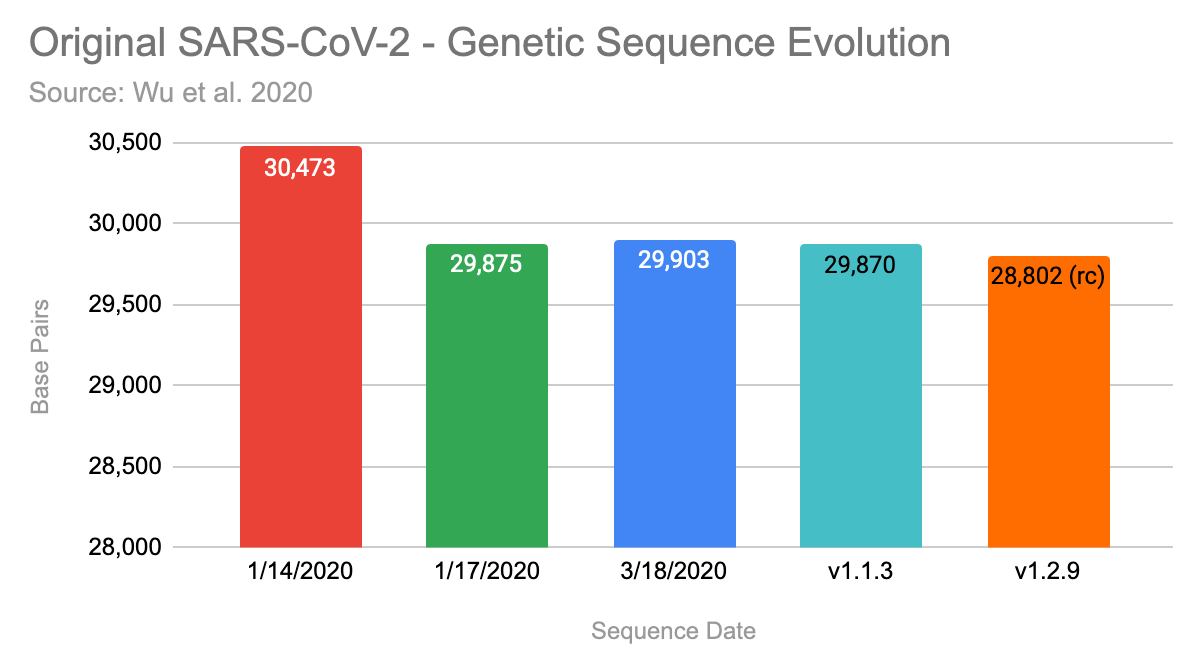
I ran Megahit 1.1.3 and 1.2.9, with the latter producing a significant shorter contig with only 29,802 bp. At least, both runs on each version produced the identical longest contig, excluding the start & end.
Finally, it is noteworthy, that none of the sequences assembled by Megahit produced the full claimed contig. Also the full genome was never assembled, as the end of the full genome is always missing.
More on this in my next article….
Please let me know your thoughts and feedback in the comments, and don’t forget to share this article if you liked it!
Also check out the code: https://github.com/USMortality/Megahit-SARS-CoV-2
Also see my next article on this topic:

No virus ever proven to exist directly in the fluids of a human. Are you just learning this now?
Thanks, Ben, I listened to the Twitterspace months ago. I'm not very familiar with the details. But what I remember is that there were ~30000 contigs built in ~350000 possible. PC generated many and they took the longest version. Is that correct? Shame on the commenters in that space. Please keep going!