


How Reliable is Megahit?
A Quality Control Study of the Tools Used to Discover the SARS-CoV-2 Genome

Introduction
The Wu et al. 2020 paper is the first to discover the genetic sequence of the novel pathogen SARS-CoV-2. As I have described earlier in my genomics series, they used a combination of bioinformatics techniques, such as de-novo assembly and alignment, to construct the genome from short paired reads (~ 2x150 max) of all collected RNA from the patient sample.
Last time I showed, how artificially simulated reads from the reference genome can be perfectly aligned against the tails. Today, let’s take it a step further and do some quality control of three common aligners (bwa, bowtie2, minimap2) and the de-novo-assembler Megahit, which was used to construct the original SARS-CoV-2 reference genome.
Overview of my Seven Part Genomics Series:







Methods
I have downloaded reference genomes of 24 common viruses, including SARS1/2, Ebola, Influenza and Measles. In a first step, we’ll run each genome through the step of random fragmentation into ~100bp long reads (min: 50bp; max: 150bp), generated via my genome.ts script and Sanger institute’s wgsim tool. Then we will use these reads to align them against the reference genome, and in a second step use Megahit to assemble the reads and compare the produced assembly (contigs) to the reference genome.
Overview of genomics series
Alignment
Separate
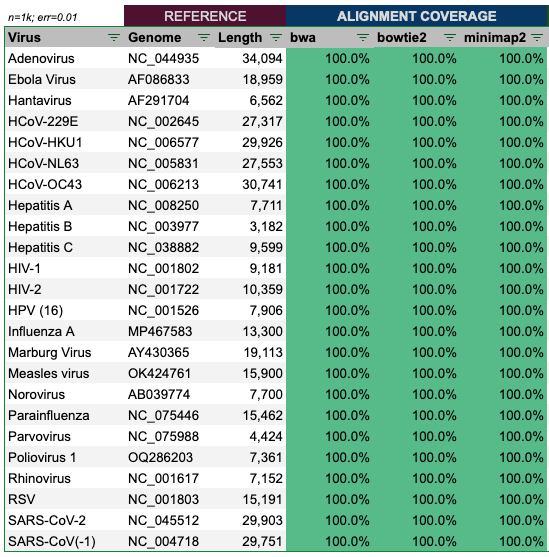
Alignment of the reads produced by genome.ts with 1,000 virion copies and error rate of 1%, show a perfect alignment in all cases.
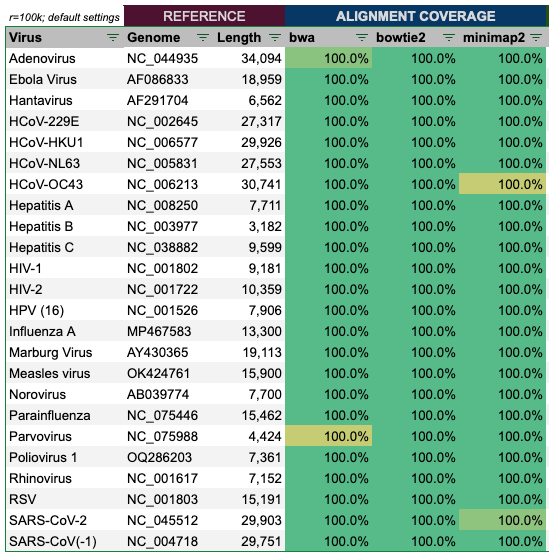
Alignment of the reads produced by wgsim with 100,000 reads and default settings, show an almost perfect alignment in all cases as well.
Some cells are slightly yellow, as they are closer to 99.999% aligned, but still pretty good, and it is maximum only 1 out of the three tools.
Mixed
In a further step, I have ‘mixed’ all reads of the 24 genomes together. Here also the result was 100% for all aligners.
Hence, we can conclude, that if at least two aligners are used, the alignment results appear to be 100% accurate.
Assembly
Separate
The next step was to test the de-novo assembler Megahit. I used the same procedure to generate the random reads, both with genome.ts(1) and wgsim(2).
First, I only used random reads of each genome and let Megahit (re)-assemble the reads into contigs, which I then compared against the reference genome with blast. Here are the results:

To my surprise, Megahit was not able to perfectly assemble the reads for most genomes. Only 7 of 24 (1) and 5 of 24 (2) resulted in an identically assembled contig. In combination of the two methods, Megahit could only reliably (re)-assemble 2 of 24, that’s 8% of the genomes. When setting the bar a bit lower at 99.9% completeness, 75% of genomes were complete, and at the 99% level 83% of genomes were complete. In the case of the reads generated for Parvovirus, HPV and Measles the reads generated by wgsim with default settings likely added an extra base, which resulted in a longer than expected contig.
Mixed
The real test however is mixing all reads of the 24 genomes, and letting Megahit (re)-assemble. Here the results look even worse:
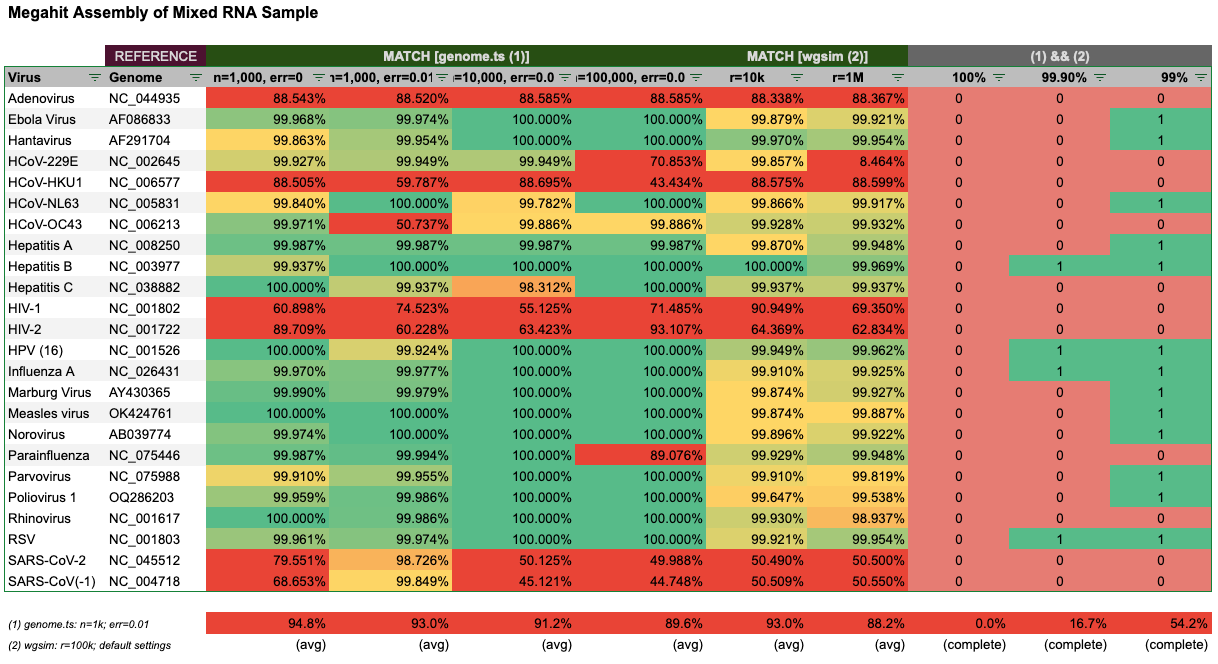
Depending on how many virions are assumed to be in the sample (n) the results vary, but we can see that even at 100,000 virions, Megahit failed to assemble between 4–8 genomes to a significant length.
It’s especially surprising that Megahit failed to assemble SARS-1 and SARS-2 fairly consistently. Overall, Megahit did not reliably assemble 100% of the genomes in any case. At 99% completeness, only 4 genomes could be reliably recovered and at 99% about half of the genomes.
I have also mixed in the original Wu et al. 2020 reads back into the mix, with no significant difference in outcome. However, a real world test would be to mix in unknown RNA from non-viral sources for true quality assessment. Since Megahit already performed poorly on this ideal setup, a better result is not to be expected.
Summary
Read alignment appears to be accurate. However, as alignment has to be carried out against a reference genome, which is typically identified at first via de-novo assembly, the reliability of the assembly step should be even more important. Here, the de-novo assembly with Megahit - which was used to discover the SARS-CoV-2 genome - shows insufficient reliability in reconstructing entire genomes, as it has failed to reconstruct between 4-8 of the 24 tested reference genomes (17%-33%), including SARS-CoV(-1) and SARS-CoV-2.
WOW
The overlords don’t want billions of people using up the earths resources. Satan wants to modify the natural God-given genome into his own image.
The intercession will occur in time to preserve a remnant of the natural genome.